
Гормоны жировой ткани и их роль в патогенезе сахарного диабета 2-го типа
М. И. Балаболкин, доктор медицинских наук, профессор
Е. М. Клебанова, доктор медицинских наук
Первый МГМУ им. И. М. Сеченова, Москва
В последние 30–40 лет отмечается значительное увеличение заболеваемости сахарным диабетом (СД) во всем мире и особенно в промышленно развитых странах, где 6–10% населения страдают этим заболеванием и его распространенность имеет четкую тенденцию к увеличению, в первую очередь, в возрастных группах старше 40 лет. Каждые 15 лет число больных сахарным диабетом удваивается. Это происходит в основном за счет прироста лиц, страдающих СД 2-го типа.
По данным ВОЗ, в 2003 году в мире насчитывалось около 180 млн больных СД. К настоящему времени их количество превышает 200 млн. Согласно экспертной оценке, к 2010 г. в мире будет насчитываться более чем 230 млн, а к 2025 г. — 300 млн больных СД, из которых 80–90% будут составлять больные СД 2-го типа.
Большая социальная значимость СД состоит в том, что он приводит к ранней инвалидизации и летальности, которая обусловлена наличием поздних сосудистых осложнений диабета: микроангиопатии (ретинопатия и нефропатия), макроангиопатии (инфаркт миокарда, инсульт, гангрена нижних конечностей), нейропатии. СД очень часто становится причиной слепоты, смерти от уремии. У таких больных наиболее велик риск развития сердечно-сосудистых заболеваний. Более 40% всех не обусловленных травмой ампутаций нижних конечностей проводится в связи с синдромом диабетической стопы и гангреной нижних конечностей. Дистальная полинейропатия и автономная нейропатия снижают качество жизни больных, приводя к нарушению трудоспособности и инвалидизации, и нередко являются причиной летальных исходов. Естественное течение СД 2-го типа, развитие сосудистых осложнений и их исход представлены на рис. 1.
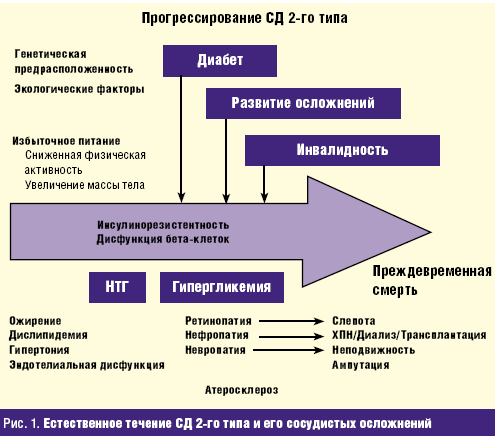
Эссенциальный СД 2-го типа является гетерогенным и полигенным заболеванием, в патогенезе которого участвуют несколько генетических и внешнесредовых компонентов. Взаимосвязь наследственных факторов и факторов внешней среды в прогрессировании нарушений углеводного обмена при СД 2-го типа представлена на рис. 2.

Как видно из данных, представленных на рис. 2, основными факторами патогенеза СД 2-го типа являются инсулиновая резистентность и недостаточность функции бета-клеток, которые развиваются, как правило, лишь через значительное время от начала клинической манифестации заболевания. Инициирующим моментом в патогенезе СД 2-го типа является взаимодействие генетических и внешних компонентов. Гены, определяющие предрасположенность к СД 2-го типа, работают уже на самых ранних (эмбриональных) стадиях развития поджелудочной железы и оказываются вовлечены в процессы секреции инсулина и обмена глюкозы в бета-клетке, печени и в других тканях организма.
Наследование СД 2-го типа полигенное, в качестве генов-кандидатов рассматриваются следующие гены: ген инсулина, ген рецептора к глюкагону, ген белка, связывающего свободные жирные кислоты, ген гликогенсинтазы, ген белковой фосфатазы типа 1, ген фратаксина, гены глюкозных транспортеров (ГЛЮТ-2 и ГЛЮТ-4), ген бета3-адренорецептора, ген гексокиназы типа 2, ген фосфатидилинозитол-3-киназы, гены прогормональной конвертазы и карбоксипептидазы Е, ген амилина, ген рецептора желудочного ингибиторного полипептида, ген островка-1, ген рецептора глюкагон-подобного пептида типа 1, ген RAD, ген рецептора витамина D, ген белка, связывающего витамин D, ген промотора глюкозо-6-фосфатазы, ген промотора фосфоэнолпируваткарбоксилазы и ген инсулинрезистентного СД 2-го типа, локализованный на длинном плече 20-й хромосомы — локус 20 q13.1–13.2.
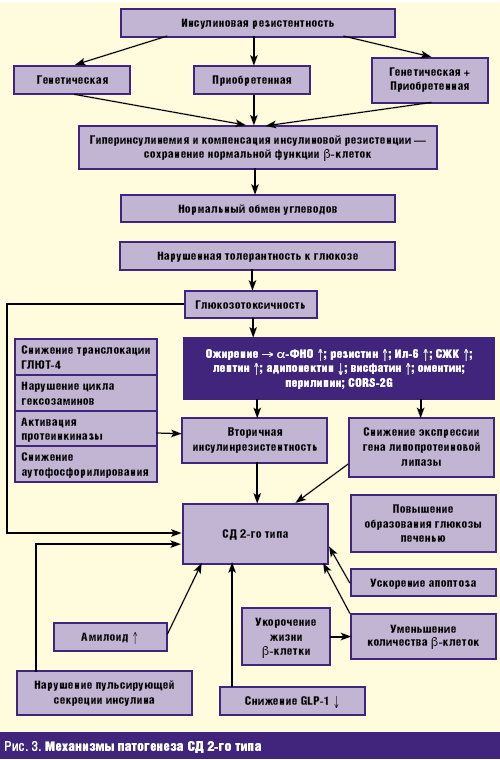
Указанные гены осуществляют свое влияние в кооперации с генами, вовлеченными в патогенез ожирения. Если мутация одних генов четко ассоциируется с СД, то мутация других остается как бы «молчащей» и не сочетается с известными нам клиническими и биохимическими признаками заболевания. Более того, мутации генов, сочетающиеся с СД 2-го типа, неодинаково проявляют себя в различных популяциях, что, видимо, связано с пока неизвестными нам факторами, которые реализуют имеющуюся мутацию генов в определенные нарушения функции отдельных органов и систем, приводя к развитию определенной клинической картины заболевания. Механизмы развития патогенеза СД 2-го типа представлены на рис. 3.
У 90–95% больных, страдающих СД 2-го типа, имеется различная степень ожирения. Жировая ткань является основным «хранилищем» запасов энергии в организме. Из всей энергии, поступающей в организм с пищей, около 75% расходуется на поддержание основного обмена, около 10–15% от ее количества используется в процессе работы и при различных формах физической активности и 10–15% — на поддержание постоянной температуры тела, т. е. термогенез. Помимо избыточного количества жировой ткани риском для развития диабета является ее распределение, т. е. тип ожирения. Преимущественное отложение жировой ткани в большом сальнике и ретроперитонеальном пространстве характерно для «абдоминального», или «андроидного», типа ожирения, при котором фигура приобретает форму яблока.
Преимущественное отложение жировой ткани в нижней части туловища и бедер характерно для женского типа ожирения, при котором фигура приобретает форму груши. Абдоминальный тип ожирения сочетается с СД 2-го типа в комплексе с дислипидемией, сердечно-сосудистыми нарушениями, гипертензией. При абдоминальном типе ожирения у женщин выявляется повышенное содержание в сыворотке крови андрогенов и кортизола, на фоне снижения глобулина, связывающего половые гормоны. Развитие такого типа ожирения усугубляется такими вредными привычками, как курение и потребление алкоголя. Абдоминальный тип ожирения чаще сочетается с СД 2-го типа. Во-первых, в абдоминальных жировых депо скорость липолиза значительно выше, чем в подкожно-жировой клетчатке, и свободные жирные кислоты, высвобождаемые в период липолиза, по системе воротной вены непосредственно поступают в печень, приводя к повышению синтеза липопротеинов и их чрезмерному поступлению в эндотелиальные и мышечные клетки.
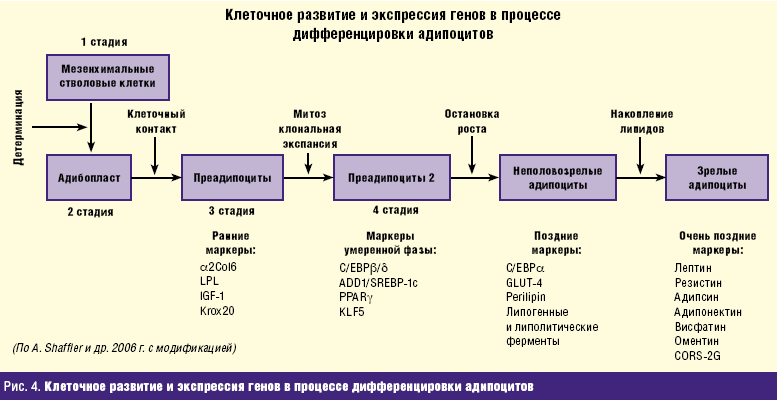
Как показали исследования последних лет, жировая ткань является также эндокринной железой, секретирующей значительное количество гормонов и биологически активных пептидов, к которым относятся: лептин, пантофизин, резистин, фактор некроза опухолей-бета (ФНО-бета), адипонектин, висфатин, внутриадипоцитные альтернативные белки (адипсин, С3, В), внутриадипоцитный белок 30 kD (ACRP30), белок, стимулирующий ацетилирование (ASP), липопротеиновая липаза (LPL), белок, переносящий эфиры холестерина, аполипопротеин Е (Apo E), белок, связывающий ретинол, сосудистый эндотелиальный фактор роста (VEGF), интерлейкин-6, ангиотензиноген, ингибитор 1 типа активатора плазминогена (PAI-1), трансформирующий фактор роста-бета (TGF-бета), фактор роста гепатоцитов, инсулиноподобный фактор роста-1 (IGF-1), монобутирин, белки 1, 2 и 3 типа, разобщающие окислительное фосфорилирование, синтез индуцированного NO, который повышает уровень свободных жирных кислот (СЖК), инсулинорезистентность и гипертриглицеридемию, простациклин (PgI2), белки острой фазы (гаптоглобин, альфа1-кислый гликопротеин), белки внеклеточного матрикса (коллаген 1, 3, 4 и 6 типа, фибронектин; остеонектин; ламинин; матриксные металлопротеиназы 2 и 9 типа), эстрогены (р450-ароматаза конвертирует андростендион в эстрон), 17-бета-гидроксистероидная оксидоредуктаза, аgouti сигнальный белок и др., большинство из которых влияют на повышение степени выраженности инсулиновой резистентности. Клеточное развитие и экспрессия генов в процессе дифференцировки адипоцитов представлены на рис. 4.
Значительное место в развитии и поддержании инсулиновой резистентности отводится гормонам жировой ткани (рис. 5).
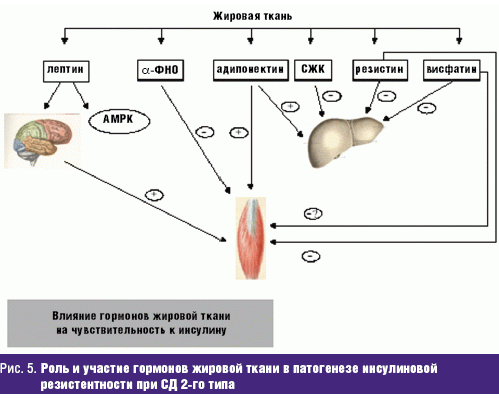
Гормоны жировой ткани, за исключением адипонектина, снижают чувствительность периферических тканей к инсулину, что сопровождается повышением степени выраженности инсулиновой резистентности, которая участвует и является основным звеном в патогенезе СД 2-го типа. Наряду с этим влияние адипонектина на состояние инсулиновой резистентности противоположно действию других гормонов жировой ткани. Секреция этого гормона снижена при СД 2-го типа, а ее восстановление сопровождается улучшением углеводного обмена при СД, снижением атерогенеза и замедлением прогрессирования сосудистых осложнений диабета.
Известно, что инсулиновая резистентность при СД 2-го типа более выражена у больных, страдающих абдоминальным или висцеральным типом ожирения. Оказалось, что эти различия обусловлены неодинаковой экспрессией генов гормонов жировой ткани в абдоминальной и подкожной жировой клетчатке. Данные об экспрессии генов гормонов жировой ткани представлены в табл.
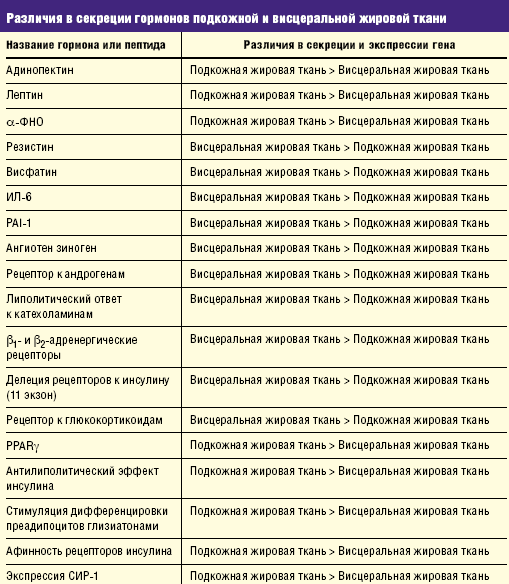
Данные, представленные в табл., показывают, что висцеральная или абдоминальная жировая клетчатка секретирует значительно более высокое количество гормонов, усиливающих проявление инсулиновой резистентности (резистин и др.), причем в висцеральной клетчатке снижается секреция гормонов (адипонектин), влияние которых заключается в снижении степени выраженности инсулиновой резистентности и замедлении прогрессирования сосудистых осложнений диабета.
Лептин — белок с мол. м. 16 kDa — секретируется в основном в жировой ткани, хотя небольшое его количество образуется также в мышцах и плаценте. В системном кровообращении он присутствует в «свободной» и «связанной» с белками плазмы форме. Его клиренс происходит в основном в почках. При голодании секреция лептина уменьшается, а при переедании и ожирении — увеличивается. Физиологическая функция лептина заключается, вероятнее всего, в предупреждении развития ожирения в условиях избыточного поступления пищи в организм. Снижение секреции лептина при голодании является своего рода сигналом для повышения поглощения энергии. При избыточном поступлении пищи в организм повышается, с одной стороны, термогенез, путем активирования образования энергии в буром жире, посредством индукции экспрессии генов, ответственных за синтез так называемых митохондриальных разобщающих окислительное фосфорилирование белков 1, 2 и 3 типа, которые регулируют скорость термогенеза в организме.
Снижение уровня лептина в крови ниже порогового уровня сопровождается повышением аппетита, а изменение секреции гипофизарных гормонов характеризуется теми же параметрами, как это имеет место при голодании. Гиполептинемия увеличивает чувство голода и угнетает функцию репродуктивной системы. Гиперлептинемия, наблюдаемая при ожирении, не сопровождается значительными изменениями состояния здоровья и является своеобразным сигналом снижения массы жира и наличия голодания.
Переход лептина в ЦНС опосредуется рецепторами гемато-энцефалического барьера. В течение дня концентрация лептина в плазме крови флюктуирует в соответствии с приемом пищи, ее количеством и наличием количества жира в организме. В течение ночи в постабсорбционный период концентрация лептина в плазме повышается пропорционально количеству жировой ткани в организме. В аркуатном ядре гипоталамуса идентифицированы два типа клеток, один из которых отвечает за образование нейропептида Y (NPY) и agouti-подобного белка, которые являются пептидами, стимулирующими прием пищи. Лептин снижает экспрессию генов указанных белков. Образование комплексов с рецепторами лептина, локализованными на клетках второго типа аркуатного ядра гипоталамуса, вызывает повышение экспрессии кокаин- и амфетамин-подобных транскрипт и бета-меланоцитостимулирующего гормона, которые, в свою очередь, являются белками, ингибирующими прием пищи. К настоящему времени клонировано по крайней мер 6 изоформ рецептора к лептину, с помощью которых опосредуются биологические влияния гормона. Большинство известных эффектов лептина опосредуется через рецепторы LRb (рис. 6).
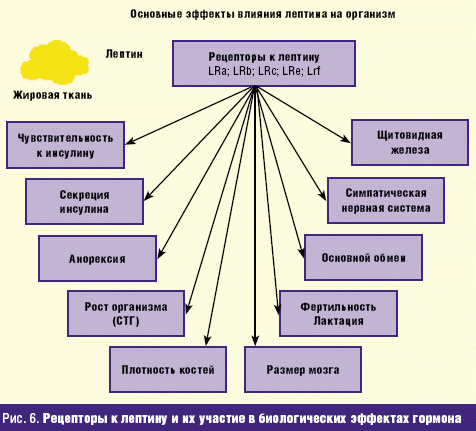
Образованный комплекс лептина с соответствующим рецептором (изоформа LRb) характеризуется широким спектром биологических эффектов: регуляция энергетического баланса в организме, участие в контроле складирования и высвобождения глюкозы как непосредственно, так и опосредованно — путем изменения чувствительности тканей к инсулину или его секреции; снижение аппетита, вплоть до анорексии; повышение основного обмена в сочетании с активацией тонуса симпатической нервной системы и функции щитовидной железы; угнетение секреции стрессовых гормонов и, в частности, глюкокортикоидов; активизация процессов роста мозга и увеличение его размеров; изменение функциональной активности системы гипоталамус–половые железы, вплоть до нарушения лактации и фертильности.
У человека врожденная недостаточность лептина сопровождается ожирением, гиперфагией и гипогонадотропным гипогонадизмом. Применение экзогенного лептина сопровождается значительным снижением аппетита, избыточной массы тела и инициирует развитие пубертата. Возможность предупреждения снижения концентрации лептина в плазме крови и снижения массы тела способствует сохранению функции щитовидной железы и скорости использования энергии в организме. Терапия рекомбинантным лептином больных ожирением без недостаточности секреции лептина приводит лишь к умеренному снижению массы тела. Заместительная терапия лептином предупреждает изменение соотношения лютеинизирующий гормон/тестостерон в плазме крови натощак. Однако при этом лептин не предупреждает изменений в уровне циркулирующих Т3 и rТ3, пульсирующей секреции стоматотропного гормона (СТГ) и кортизола (J. L. Chan и соавт., 2003).
Предположение о том, что недостаточность секреции лептина у человека сопровождается ожирением, не находит клинического подтверждения. Уровень лептина в сыворотке крови повышается с увеличением ожирения и массы тела, тогда как доказанная недостаточность секреции лептина встречается крайне редко. Эти данные позволяют считать, что при ожирении, вероятнее всего, имеет место резистентность к лептину на уровне транспорта в ЦНС или на пострецепторном уровне. Это предположение, вероятнее всего правильное, требует проведения дополнительных исследований, так как показано, что уровень лептина в плазме крови достаточно четко коррелирует с количеством жировой ткани в организме (M. D. Jensen и соавт., 1999). У больных с липоатрофиями, у которых содержание лептина в сыворотке крови снижено, терапия лептином сопровождается снижением количества принимаемой пищи и улучшением показателей метаболизма (E. A. Oral и соавт., 2003). Основным регуляторным механизмом секреции лептина является прием пищи, который сопровождается повышением секреции инсулина и лептина, тогда как голодание ассоциируется с повышением секреции контринсулиновых гормонов и снижением секреции лептина.
Два цитокина (ФНО-бета и интерлейкина-6 (ИЛ-6)), образующиеся в жировой ткани, также могут влиять на чувствительность периферических тканей к инсулину. Исследования, посвященные изучению роли провоспалительных цитокинов (ФНО-бета и ИЛ-6, а также С-реактивного белка), позволили предположить, что воспаление участвует в патогенезе инсулиновой резистентности. Считается, что хроническое субклиническое воспаление является частью синдрома инсулиновой резистентности, а указанные цитокины служат предикторами сосудистых осложнений диабета (A. Festa и соавт., 2000). ФНО-бета имеет м. м. 17 kDa, и повышение его содержания в сыворотке крови сочетается с наличием ожирения, инсулиновой резистентностью, увеличением концентрации С-реактивного белка и ИЛ-6, а также ускорением апоптоза (S. W. Coppack, 2001). ФНО-бета является цитокином, и его ген экспрессируется как в иммунных, так и в неиммунных клетках, включая эндотелиоциты, фибробласты и адипоциты. Установлено, что высвобождение ФНО-бета из клеток жировой ткани аналогично его высвобождению из моноцитов или макрофагов. Повышение экспрессии гена ФНО-бета в адипоцитах животных и при ожирении у человека сопровождается повышением степени выраженности инсулиновой резистентности. Это позволило сделать вывод, что данный цитокин является одним из ключевых медиаторов ее развития. Это антиинсулиновое действие ФНО-бета является следствием его влияния на снижение экспрессии ГЛЮТ-4 и ингибирования тирозинкиназы рецепторов к инсулину в клетках и тканях-мишенях.
Что касается ИЛ-6, то уровень экспрессии гена ИЛ-6 в жировой ткани напрямую коррелирует как со степенью активирования поглощения глюкозы, так и с выраженностью инсулиновой резистентности, что подтверждается исследованиями in vivo и in vitro (J. P. Bastard и соавт., 2002). Внутривенное введение ИЛ-6 у человека сопровождается повышением уровня СЖК и глицерола в сыворотке крови, что является следствием его влияния на липолиз жировой ткани (van Hall и соавт., 2003). Определенный интерес представляет тот факт, что в жировой ткани помимо образования ИЛ-6, который является провоспалительным цитокином, секретируется и другой цитокин-рецепторный антагонист интерлейкин-1, обладающий антивоспалительным эффектом (C. E. Juge-Aubry и соавт., 2003).
Определенную роль в патогенезе инсулиновой резистентности играет и ФНО-альфа, уровень которого в жировой ткани коррелирует с массой жировой ткани и гиперинсулинемией у мышей. Если лептин и ФНО-альфа способствуют развитию инсулиновой резистентности и их содержание в сыворотке крови и тканях напрямую коррелирует со степенью выраженности инсулиновой резистентности, то концентрация адипонектина в плазме крови характеризуется отрицательной корреляцией с инсулиновой резистентностью. Это позволяет считать циркулирующий уровень адипонектина маркером инсулиновой резистентности и риска развития ангиопатий. Более того, содержание адипонектина в сыворотке крови может служить объективным маркером снижения риска развития ишемической болезни сердца (ИБС) как у больных СД 2-го типа, так и у больных с нарушенной гликемией натощак (H. Knobler и соавт., 2006). Knobler и соавт., проводившие длительное мониторирование (6,2 ± 1,3 года) группы с нарушенной гликемией натощак, показали что у 44% (256 из 588) больных развился СД. У них на момент начала исследования были выше индекс массы тела (ИМТ), содержание гликемии натощак, С-реактивного белка, триглицеридов и индекс инсулиновой резистентности, при статистически достоверном сниженном уровне адипонектина в сыворотке крови. Эти данные еще раз подтвердили, что высокое содержание адипонектина в сыворотке крови сочетается со сниженным риском развития СД.
Проведенные исследования свидетельствуют о том, что ФНО-альфа:
- принимает участие в регуляции обмена углеводов и жиров в организме;
- действует как митогенный фактор в апоптозе адипоцитов;
- стимулирует секрецию лептина, регулирует функцию митохондрий и экспрессию генов;
- индуцирует инсулинорезистентность в жировой ткани и мышцах;
- ингибирует секрецию инсулина бета-клетками островка поджелудочной железы;
- участвует в патогенезе и прогрессировании сосудистых осложнений диабета;
- снижает экспрессию гена ГЛЮТ-4;
- ингибирует тирозинкиназу рецептора инсулина;
- повышает фосфорилирование серина в СИР-1, что сопровождается снижением функции рецептора инсулина;
- снижает экспрессию гена липопротеиновой липазы.
Жировая ткань является также местом образования еще одного гормона — адипонектина, который представляет собой полипетид с м. м. 30 kDa, содержащий 244 аминокислотных остатка, сходный по структуре с молекулой коллагена и ФНО-бета и циркулирующий в периферическом кровообращении в 8 различных изоформах. Ген адипонектина локализуется на хромосоме 3 q27. Его концентрация в крови, как и у лептина, имеет обратную корреляцию с массой жировой ткани и степенью выраженности инсулиновой резистентности. Снижение уровня адипонетина в сыворотке крови имеет место при СД 2-го типа и ИБС. На основании этих наблюдений было сделано предположение, что адипонектин повышает чувствительность тканей к инсулину и обладает кардиопротективными эффектами (J. J. Diez и P. Iglesias, 2003).
Адипонектин оказывает биологический эффект посредством связывания с рецепторами двух типов (T. Yamauchi и соавт., 2003), активирование которых сопровождается снижением массы тела без уменьшения приема пищи, увеличением окисления жирных кислот в скелетных мышцах и печени, а также снижением их уровня в сыворотке крови. Кроме того, наблюдается уменьшение содержания глюкозы в крови без увеличения секреции инсулина, а также снижение содержания триглицеридов в мышцах и печени, что свидетельствует о повышении чувствительности тканей к инсулину и снижении инсулиновой резистентности. Наблюдаемое под влиянием адипонектина уменьшение экспрессии адгезивных молекул эндотелиальными клетками сосудов и скорости образования количества цитокинов макрофагами позволяет считать, что адипонектин относится к антиатеротогенным эндогенным соединениям.
Сенситайзеры инсулина (Актос и Авандия), применяемые в настоящее время для лечения СД 2-го типа, повышают содержание адипонектина в сыворотке крови больных, что благоприятно влияет на течение диабетических ангиопатий. Установлено, что повышение инсулиновой резистентости, наблюдаемое при применении глюкокортикоидов, бета-адренергических агонистов и ФНО-бета, является следствием их ингибирующего влияния на образование адипонектина.
Содержание адипонектина в сыворотке крови имеет обратную корреляцию с триглицеридами, атерогенным индексом, АпоВ или АпоЕ и положительную корреляцию с холестерином липопротеидов высокой плотности (ЛПВП) и уровнем АпоА-1. Адипонектин повышает чувствительность периферических тканей к инсулину, увеличивает окисление жира на периферии, снижает уровень СЖК в крови, уменьшая внутриклеточное содержание триглицеридов в печени и мышцах. Помимо этого, адипонектин ингибирует экспрессию адгезивных молекул в эндотелиальных клетках и образование цитокинов макрофагами, следствием чего является угнетение воспалительных процессов.
Адипонектин, таким образом, повышает чувствительность тканей к инсулину и обладает антивоспалительными и антиатерогенными свойствами, а высвобождаясь в кровеносную систему, он накапливается в сосудистой стенке в ответ на повреждение эндотелия и модулирует воспалительный процесс в эндотелии.
Резистин, или адипоцит-специфический секреторный фактор (ADSF/FIZZ3), является пептидом, состоящим из 114 аминокислотных остатков. Ген резистина локализуется у человека на хромосоме 19 р13.3. Резистин принадлежит к семейству цистеин-содержащих С-терминальных доменовых белков, называемых резистин-подобными (RELM) или FIZZ молекулами, вовлеченными в процессы воспаления. Резистин секретируется как преадипоцитами, так и адипоцитами. Кроме того, в период эмбрионального развития ген резистина экспрессируется трофобластами плаценты преимущественно в конце беременности, и его содержание в плазме крови беременных женщин значительно выше. Считается, что в этот период резистин выполняет роль регулятора углеводного обмена. Установлено, что повышенная экспрессия гена резистина в жировой ткани человека при центральном (абдоминальном) ожирении коррелирует с наличием СД 2-го типа сердечно-сосудистых заболеваний.
Полиморфизм гена резистина (3’UTR + 62G → A) идентифицирован у больных, страдающих СД 2-го типа и относящихся к китайской популяции. Тиазолидиндионы снижают экспрессию гена резистина, чем и объясняется эффект препаратов указанной группы на уменьшение степени выраженности инсулиновой резистентности (C. M. Steppan и соавт., 2001). Изучение биологического действия резистина, секретируемого адипоцитами и эндокринными клетками желудочно-кишечного тракта, показало, что резистин индуцирует печеночную, но не периферическую резистентность к инсулину у крыс и, таким образом, отвечает за повышение скорости образования глюкозы печенью (M. W. Rajala и соавт., 2003).
Следует отметить, что жировая ткань является местом секреции и других биологически активных веществ, к которым относятся белок, стимулирующий ацетилирование (БСА (бычий сывороточный альбумин) или ASP или C3 adesАrg), и ингибитор 1 типа активатора плазминогена (PAI-1). Что касается БСА, то он является липогенным адипоцитокином и представляет собой комплекс, состоящий из компонентов альтернативного пути образования факторов комплемента. Считается, что белок, стимулирующий ацетилирование, образуется в результате взаимодействия нескольких факторов комплемента, таких как фактор С3, фактор В и фактор D (адипсин или БСА). Роль и биологическое значение БСА интенсивно изучаются. Показано, что липопротеины и, в частности, уровень хиломикронов вызывают увеличение высвобождения БСА.
У человека содержание БСА в сыворотке крови имеет обратную корреляционную зависимость с распределением глюкозы в организме в условиях эугликемической клэмп-методики (P. J. Havel, 2002). Это может свидетельствовать о его роли в формировании чувствительности тканей к инсулину. БСА вовлечен в обмен жиров (ингибирует гормон-чувствительную липазу, повышает активность диацилглицеринтрансферазы и эстерификацию жирных кислот, синтез триглицеридов, увеличивая их депонирование в жировых депо) и углеводов (увеличивает поглощение глюкозы периферическими тканями и ускоряет транслокацию глюкозных транспортеров к периферии клетки). Хотя эти эффекты проявляются независимо, они дополняют действие инсулина. Концентрация белка, стимулирующего ацетилирование в сыворотке крови, повышена у больных, страдающих ожирением, СД 2-го типа и ИБС.
В последнее время идентифицирован еще один гормон жировой ткани — висфатин, ген которого экспрессируется в висцеральном жире и способствует его дальнейшему накоплению. Не исключено, что висфатин оказывает свое биологическое действие не только через специфические рецепторы, но и через инсулиновые рецепторы. мРНК висфатина определяется в моноядерных клетках крови у больных СД 2-го типа, и ее количество в несколько раз выше у больных СД 2-го типа по сравнению с пациентами с диабетом, имеющими дефицит веса, или практически здоровыми лицами. Уровень висфатина в циркулирующих клетках крови напрямую коррелирует с ИМТ, окружностью талии и индексом инсулиновой резистентности. Считается, что висфатин участвует в патогенезе сосудистых осложнений диабета и атерогенезе.
Таким образом, гормоны жировой ткани оказывают как прямое, так и опосредованное влияние на процессы патогенеза СД 2-го типа и развитие сосудистых осложнений. В связи с этим медикаментозное воздействие на угнетение секреции гормонов жировой ткани, участвующих в повышении степени выраженности инсулиновой резистентности, или восстановление до нормальных значений содержания адипонектина в сыворотке крови будут способствовать лучшей компенсации углеводного обмена при СД и профилактике развития его сосудистых осложнений.
Литература
- Schaffler A., Muller-Ladner U., Scholmerich J., Buchler C. Role of adipose tissue as an inflammatory organ in human diseases // Endocr Rev. 2006. Vol. 27. P. 449–467.
- Chan J. L., Hest K., DePaoli A. M. et al. The role of falling leptin levels in the neuroendocrinology and metabolic adaptation to short-term starvation in healthy men // J Clin Invest. 2003. Vol. 111. P. 1409–1421.
- Jensen M. D., Hensrud D. D., O’Brien P. C. et al. Correlation and interpretation of plasma leptin concentration data in humans // Obes Res. 1999. Vol. 7. P. 241–245.
- Oral E. A., Simha V., Ruiz E. et al. Leptin-replacement therapy for lipodystrophy // New Engl J Med. 2003. Vol. 346. P. 570–578.
- Festa A., D’Agostino R., Howard G. et al. Chronic subclinical inflammation as part of the insulin resistance syndrome: the Insulin Resistance Atherosclerosis Study (IRAS) // Circulation. 2000. Vol. 102. P. 42–47.
- Coppack S. W. Pro-inflammatory cytokines and adipose tissue // Proc Nutr Soc. 2001. Vol. 60. P. 349–356.
- Bastard J. P., Maachi M., Van Nhieu J. T. et al. Adipose tissue Il-6 content correlates with resistance to insulin activation of glucose uptake both in vivo and in vitro // J Clin Endocrinol Metabol. 2002. Vol. 87. P. 2084–2089.
- Van Hall G., Steensberg A., Sacchetti M. et al. Interleukin-6 stimulates lipolysis and fat oxidation in humans // J Clin Endocrinol Metabol. 2003. Vol. 88. P. 3005–3010.
- Juge-Aubry C. E., Somm E., Giusti M. et al. Adipose tissue is a major source of interleukin-1 receptor antagonist: up regulation in obesity and inflammation // Diabetes. 2003. Vol. 52. P. 1104–1110.
- Knobler H., Benderly M., Boyko V. et al. Adiponectin and the development of diabetes in patients with coronary artery disease and impaired fasting glucose // Eur J Endocr. 2006. Vol. 154. P. 87–92.
- Diez J. J., Iglesias P. The role of the novel adipocyte-derived hormone adiponectin in human disease // Eur J Endocr. 2003. Vol. 148. P. 293–300.
- Yamauchi T., Kamon J., Ito Y. et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects // Nature. 2003. Vol. 423. P. 762–769.
- Steppan C. M., Bailey S. T., Bhat S. et al. The hormone resistin links obesity to diabetes // Nature. 2001. Vol. 409. 307–312.
- Rajala M. W., Obici S., Schherer P. E. et al. Adipose-derived resistin and gut-derived resistin-like molecule-b selectively impair insulin action on glucose production // J Clin Invest. 2003. Vol. 111. P. 225–230.
- Havel P. J. Control of energy homeostasis and insulin action by adipocyte hormones: leptin, acylation stimulating protein and adiponectin // Curr Opin Lipidol. 2002. Vol. 13. P. 51–59.
Статья опубликована в журнале Лечащий Врач
материал с сайта MedLinks.ru





