
Значение фактора роста фибробластов-23 у больных хронической болезнью почек – обзор современных исследований
Е. В. Шутов, доктор медицинских наук, профессор
ГБОУ ДПО РМАПО Минздравсоцразвития России,
ГУЗ ГКБ им. С. П. Боткина Департамента здравоохранения города, Москва
Нарушение минерального обмена при хронической болезни почек (ХБП) способствует развитию гиперпаратиреоза, заболеваний кости и ведет к повышению кардиоваскулярной заболеваемости и летальности. Недавно был открыт фактор роста фибробластов-23 (fibroblast growth factor-23, FGF-23) — белок, состоящий из 251 аминокислоты (молекулярная масса 32 kDa), который секретируется из остеоцитов, главным образом из остеобластов [1]. Этот белок состоит из аминоконцевой последовательности сигнального пептида (остатки 1–24), центральной последовательности (остатки 25–180) и карбоксилконцевой последовательности (остатки 181–251). Период полужизни FGF-23 в циркуляции у здоровых людей составляет 58 мин [2]. FGF-23 проявляет свои биологические эффекты через активацию FGF-рецепторов. FGF1с-рецепторы, связываясь с Kлото (англ. Klotho) белком, становятся в 1000 раз более чувствительными для взаимодействия с FGF-23, чем другие FGF-рецепторы или Клото-белок отдельно. Белок Kлото — это 130 kDa трансмембранный белок, бета-глюкорозонидаза, который был открыт в 1997 г. M. Kuro-o. Белок Клото был назван в честь одной из трех греческих богинь судьбы — Клото, прядущей нить жизни и определяющей ее срок. Было обнаружено, что уровень белка Клото в организме с возрастом существенно снижается. Затем ученые доказали его роль в регуляции механизмов старения. Генетически модифицированные мыши, в организме которых уровень белка Клото был повышен в течение всей жизни, жили на треть дольше своих диких собратьев. Мыши с дефицитом белка Клото быстро старели, и у них стремительно развивался атеросклероз и кальциноз. Белок Клото представляет собой тот редчайший случай в биологии млекопитающих, когда один-единственный белок столь существенным образом влияет на продолжительность жизни и связанные с этим физиологические процессы. Как правило, такие сложные процессы регулируются множеством генов, и роль каждого из них сравнительно невелика.
Роль FGF-23 в метаболизме фосфора
Биологическая активность и физиологическая роль FGF-23 была выяснена только в последнее время. На моделях животных (нокаутных мышах по FGF-23) было показано повышение реабсорбции фосфора (Р) и уровня 1,25-дигидрооксивитамина D (1,25 (ОН)2D) [3, 4]. Мыши с отсутствием FGF-23 характеризовались тяжелой кальцификацией сосудов и мягких тканей [5]. Важно знать, что и у мышей с отсутствием Клото-белка также отмечалась тяжелая сосудистая кальцификация, ассоциированная с гиперфосфатемией и гипервитаминозом D. Биологическая функция FGF-23 была изучена на моделях мышей при назначении рекомбинантного FGF-23 и с гиперэкспрессией FGF-23. В почках FGF-23 индуцирует фосфатурию, супрессируя экспрессию натрий-фосфорного котранспортера типа IIа и IIс в проксимальных канальцах [6, 7]. Фосфатурическое действие FGF-23 не проявляется в отсутствие натрий-водородного обменного регуляторного фактора 1 (NHERF-1) и увеличивается в присутствии паратгормона (ПТГ). Кроме того, FGF-23 супрессирует образование 1,25 (ОН)2D, ингибируя 1-альфа-гидроксилазу (CYP27B1), которая конвертирует 25-гидроксивитамин D [25 (ОН)D] в 1,25 (ОН)2D и стимулирует образование 24-гидроксилазы (CYP24), которая конвертирует 1,25 (ОН)2D в неактивные метаболиты в проксимальных канальцах почек. FGF-23 также ингибирует экспрессию интестинального натрий-фосфорного транспортера NPT2b [8], уменьшая всасывание фосфора в кишечнике. Механизм снижения уровня фосфора в крови представлен на рис. 1.
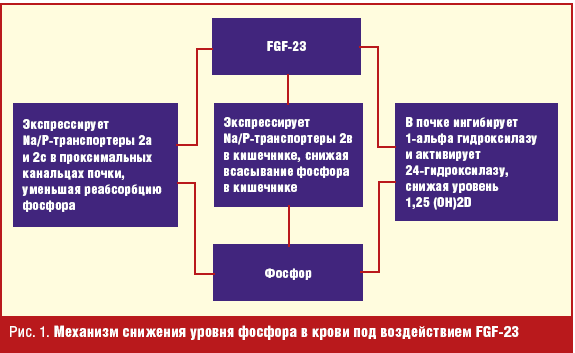
FGF-23 прямо воздействует на паращитовидные железы, регулируя секрецию и синтез паратгормона. Было показано, что FGF-23 активирует митоген-активированный протеин-киназный путь и таким образом снижает экспрессию гена ПТГ и секрецию как in vivo у крыс, так и in vitro в культуре паращитовидных клеток [9]. В другом исследовании было показано, что FGF-23 повышает экспрессию паратиреоидной 1-альфа-гидроксилазы [10], которая конвертирует 25-гидроксивитамин D [25 (ОН)D] в 1,25 (ОН)2D.
Регуляция FGF-23
Секреция FGF-23 регулируется местно в костях при участии белкового матрикса дентина-1 и фосфат-регулирующей эндопептидазы [11]. Увеличение секреции FGF-23 под воздействием 1,25 (ОН)2D показано как in vivo, так и in vitro, этот эффект опосредован через витамин D ответственные частицы, представленные в FGF-23 активаторе [12]. В клинических исследованиях показано, что назначение 1,25 (ОН)2D диализным пациентам приводило к повышению уровня FGF-23 в крови [13]. Применение высокофосфорной диеты в течение нескольких дней в экспериментальных и клинических исследованиях также приводило к увеличению уровня FGF-23 у мышей и у людей [14]. Недавно проведенные исследования показали, что эстрогены и применение парентерального железа при лечении железодефицитной анемии могут приводить к значительному повышению FGF-23 [15, 16].
FGF-23 и хроническая почечная недостаточность
Изучение уровня FGF-23 у больных с хронической почечной недостаточностью (ХПН) показало четкую его зависимость от уровня клубочковой фильтрации [17]. Повышение FGF-23 уже на ранних стадиях ХПН направлено на поддержание нейтрального баланса фосфора, за счет увеличения экскреции фосфора с мочой, уменьшения гастроинтестинальной абсорбции фосфора и супрессии продукции 1,25 (ОН)2D [18, 19]. У больных с терминальной стадией ХПН уровень FGF-23 может повышаться уже в 1000 раз по сравнению с нормой [20]. Несмотря на такое значительное повышение уровня FGF-23, оно не приводит к должному результату, что связано с дефицитом необходимого кофактора — белка Kлото, снижение уровня которого было показано в работах Koh N. с соавт. и Imanishi Y. у больных с ХПН [21, 22]. Кроме этого, повышение уровня FGF-23 происходит компенсаторно, в силу значительного снижения числа функционирующих нефронов у больных с уремией. Лечение кальцитриолом вторичного гиперпаратиреоза также может быть одной из причин повышенного уровня FGF-23, независимо от уровня фосфора в крови [23, 24]. Имеется обратная зависимость между уровнями 1,25 (ОН)2D и FGF-23 в сыворотке крови больных. Повышение FGF-23 у больных с ХПН, направленное на поддержание нормального уровня фосфора, приводит к снижению продукции 1,25 (ОН)2D, что запускает развитие вторичного гиперпаратиреоза. Паратгормон также поддерживает нормальный баланс фосфора, но не только через экскрецию фосфора, но и редуцируя экскрецию кальция и стимулируя продукцию 1,25 (ОН)2D. Однако, несмотря на это, при ХПН, в связи с уменьшением числа нефронов, компенсаторно увеличивается уровень ПТГ. При ХПН уровень FGF-23 прямо коррелирует с уровнем ПТГ, в отличие от нормы, когда имеется обратная зависимость, так как FGF-23 супрессирует синтез и экскрецию ПТГ. Это может происходить только при наличии резистентности паращитовидных желез к действию FGF-23. Подобный парадокс наблюдается и при рефрактерном вторичном гиперпаратиреоидизме, при котором нет ответа паращитовидных желез на прием кальция и кальцитриола. Это явление частично объясняется снижением экспрессии кальций-чувствительных рецепторов (CаЧР) и витамин D-рецепторов (ВДР), в паращитовидных железах с нодулярной и тотальной гиперплазией [25–27]. Недавно было также показано, что содержание белка Клото и экспрессия FGF рецепторов 1 значительно снижено при уремической гиперплазии паращитовидных желез [29, 30]. Это положение подтверждено в эксперименте на уремических крысах in vivo, когда высокое содержание FGF-23 не привело к ингибиции секреции ПТГ [31], и in vitro на культуре паращитовидных желез крыс [32]. Надо отметить, что уровень FGF-23 может быть предиктором эффективности лечения вторичного гиперпаратиреоза у диализных больных активными метаболитами витамина D [33, 34]. Длительное применение больших доз активных метаболитов витамина D при вторичном гиперпаратиреозе неуклонно ведет к повышению уровня FGF-23, а следовательно, к гиперплазии паращитовидных желез и резистентности к терапии.
FGF-23 как самостоятельный фактор риска
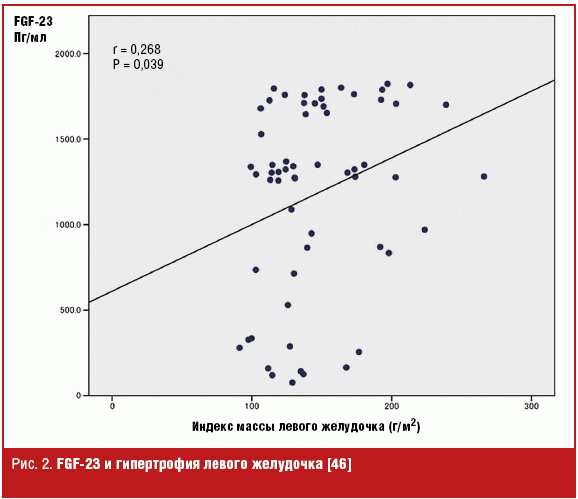
Гиперфосфатемия — один из основных факторов риска кардиоваскулярных болезней, нарушений минерального обмена и заболеваний кости. На ранних стадиях ХПН уровень фосфора поддерживается на нормальном уровне, в частности, за счет гиперсекреции FGF-23. Однако в последующем в силу ряда причин, описанных выше, наступает гиперфосфатемия, несмотря на высокий уровень FGF-23. Гиперфосфатемия прямо коррелирует с кальцификацией сосудов, кардиомиопатией, что может объяснять прямую корреляцию между уровнем фосфора, кардиоваскулярной заболеваемостью и летальностью. При высоком уровне фосфора в крови наблюдается и высокий уровень FGF-23 у больных с терминальной ХПН, этот факт мог бы отражать вторичность влияния FGF-23 на летальность. Однако недавно были получены данные, свидетельствующие о том, что летальность у больных на диализе прямо коррелирует с уровнем FGF-23, независимо от уровня концентрации фосфора в крови [35]. Одним из объяснений высокой смертности пациентов при повышении уровня FGF-23 может служить выявленная независимая ассоциация FGF-23 с гипертрофией левого желудочка (рис. 2) [36–38, 46]. Однако до последнего времени не был выяснен вопрос: FGF-23 — только простой маркер гипертрофии левого желудочка (ГЛЖ) или имеется патогенетическая связь между ними. В фундаментальной работе Christian Faul с большим авторским коллективом [39] было убедительно показано, что FGF-23 может прямо приводить к развитию гипертрофии левого желудочка. Исследование включало несколько этапов, на первом этапе было обследовано более 3000 пациентов с почечной недостаточностью, у которых определяли базовый уровень FGF-23 и проводили эхокардиографию (ЭхоКГ) через 1 год. Средний индекс массы ЛЖ (ИМЛЖ) к росту составил 52 ± 0,3 гм-2,7 (нормальный уровень < 50 у мужчин; < 47 у женщин), ГЛЖ была выявлена у 52% пациентов. Каждое увеличение на 1 логарифмическую единицу FGF-23 (lnFGF23) ассоциировалось с повышением ИМЛЖ на 1,5 г/м2 (p < 0,001), после коррекции на другие факторы риска. Затем исследователи изучили риск появления ГЛЖ у 411 пациентов, которые имели нормальные ЭхоКГ- показатели, через 2,9 ± 0,5 г. У 84 пациентов (20%) впервые была выявлена ГЛЖ, причем у нормотензивных пациентов каждое повышение на 1 ед. lnFGF23 приводило к учащению возникновения ГЛЖ de novo в 4,4 раза (p = 0,001), а высокие содержание FGF-23 обуславливало 7-кратное увеличение частоты ГЛЖ независимо от наличия или отсутствия артериальной гипертензии. В этой же работе была подтверждена гипотеза прямого влияния FGF-23 на кардиомиоциты. Сравнивали ответ изолированных кардиомиоцитов новорожденных крыс путем воздействия на них FGF-23. Иммуногистохимический и морфометрический анализ кардиомиоцитов показал значительное увеличение площади их клеточной поверхности, а также повышение уровня белка альфа-актинина, свидетельствующего об увеличении саркомеров. Были обнаружены повышение экспрессии эмбриональных бета-миозиновых тяжелых цепей (МТЦ) и одновременная депрессия зрелых альфа-миозиновых тяжелых цепей при увеличении FGF-23. Такое переключение изоформ МТЦ со зрелых на эмбриональные указывает на реактивацию эмбриональной генной программы, которая ассоциируется с гипертрофией [40–42]. FGF-23 и FGF-2 также уменьшают экспрессию предсердного и мозгового натрийуретического пептида, маркеров ГЛЖ [43]. FGF-23 уменьшает экспрессию средней цепочки ацил-КoA дегидрогеназы (СЦАГ), энзима, регулирующего оксидацию жирных кислот. Гипертрофированные кардиомиоциты переключаются на энергию с жирных кислот на углеводы, что является маркером уменьшения экспрессии СЦГА [44]. FGF-23 вызывает ГЛЖ независимо от корецептора белка Клото, который экспрессируется преимущественно в почках и паращитовидных железах и отсутствует в кардиомиоцитах [45]. Биологические эффекты факторов роста фибробластов проявляются после связывания с FGF1-FGF4-рецепторами [46], при этом FGF-23 может связываться с разными изоформами FGF-рецепторов с различной степенью аффинности [47, 48]. В работе Christian Faul с соавт. был показан прогипертрофический эффект FGF-23 и FGF-2 на кардиомиоциты, который исчезал после применения ингибитора FGF-рецепторов PD173074, что доказало возможность воздействия FGF-23 через FGF-рецепторы, независимо от белка Клото. Активация рецепторов, как было выяснено, происходит через активацию кальцийнерин-А дефосфорилирующие факторы транскрипции ядерного фактора, активирующего Т-клетки, ведущих к ядерной транслокации, а блокада их приводит к снижению действия FGF-23. Интересно отметить, что применение PD173074 предотвращало развитие ГЛЖ у крыс, несмотря на наличие у них ХПН и гипертензии.
Другой важной причиной летальности больных с ХПН является наличие у больных кальцификации сосудов, которая ассоциируется с высокой смертностью [49]. Особенно это важно с учетом большой распространенности кальцификации коронарных сосудов у диализной популяции больных (рис. 3) [50, 58].
У больных с ХПН развивается преимущественно кальцификации медии, которая ведет к повышению жесткости сосудов и высокой смертности от кардиоваскулярных причин [51]. У диализных пациентов имеются разнообразные факторы риска развития сосудистой кальцификации (уремические токсины, сахарный диабет, длительный диализ, воспаление), однако нарушение минерального обмена играет ключевую роль в этом процессе. Повышение уровня фосфора > 2,4 ммоль/л индуцирует кальцификацию гладко- мышечных клеток (ГМК) in vitro [52]. Фосфор транспортируется в клетки из экстрацеллюлярного пространства преимущественно при помощи мембранного натрий-зависимого котранспортера фосфатов III типа (Pit1), ассоциируясь с кальцификацией ГМК [53]. Подобно фосфору, повышение кальция (> 2,6 ммоль/л) в культуре медии приводит к минерализации и фенотипическому изменению ГМК [54] через Pit1, в результате ГМК трансформируются к остеобласт-подобные клетки [55]. В последнее время получены данные о прямой корреляционной связи уровня FGF-23 c кальцификацией сосудов [56]. Ассоциация FGF-23 с кальцификацией сосудов до сих пор не имеет ясного объяснения. Ряд авторов рассматривает FGF-23 как только биомаркер минерального нарушения при ХПН [57], так как понятна роль повышения уровня FGF-23 в ответ на повышение уровня фосфора в крови, а гиперфосфатемия доказанный фактор развития кальцификации сосудов. Однако новые данные свидетельствуют и о другой возможности воздействия FGF-23 на кальцификацию сосудов. Так, Giorgio Coen [58] и соавт. показали обратную зависимость между фетуином А и FGF-23, а между тем ранее было продемонстрировано, что фетуин А может синтезироваться остеобластами и храниться в костях [59], что может предполагать влияние FGF-23 на уровень фетуина А, который, как известно, предотвращает кальцификацию сосудов [60].
В работе Majd A. I. и соавт. [61] получены данные и о корреляции уровня FGF-23 с атеросклерозом, в ней авторы высказывают гипотезу, объясняющую это явление с повреждающим влиянием FGF-23 на эндотелий сосудов [62].
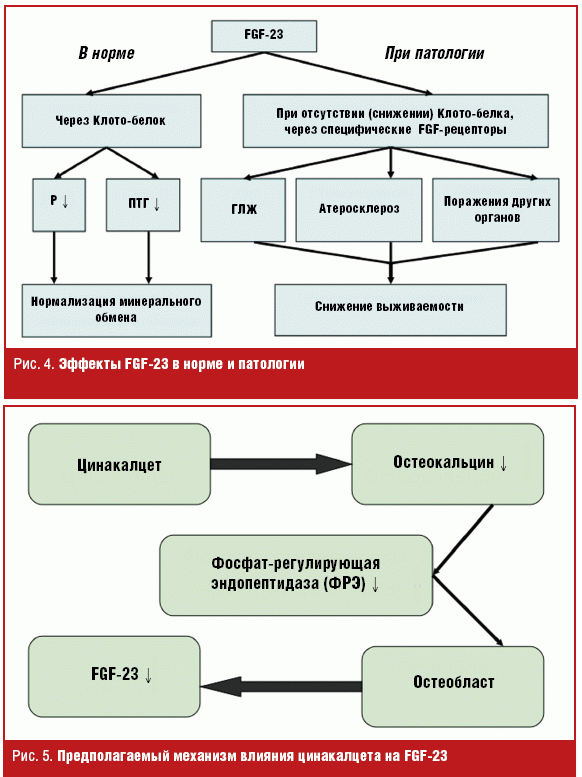
Дефицит витамина D часто наблюдается у больных с ХПН, в частности, из-за снижения продукции 1,25 (ОН)2D под влиянием FGF-23, что способствует развитию вторичного гиперпаратиреоза. Основным показанием для назначения активных метаболитов витамина D у больных с почечной недостаточностью является супрессия синтеза ПТГ и предотвращение болезней кости [63]. Однако активация витамин D-рецепторов приводит к ряду биологических эффектов: супрессии ренина [64, 65], регуляции иммунной системы и воспаления [66, 67], индукции апоптоза [68], сохранению эндотелия [69] и др. У мышей, нокаутированных по ВДР-гену, индуцируется гипертрофия и фиброз миокарда [70]. Дефицит витамина D — доказанный нетрадиционный фактор риска сердечно-сосудистых осложнений и летальности у больных с ХПН [71], но также повышает риск смерти у больных сердечной недостаточностью [72]. Кроме того, дефицит витамина D ассоциируется с сердечной недостаточностью и внезапной смертью в общей популяции [73, 74]. Высокий уровень FGF-23 ассоциируется с низким содержанием витамина D, что также может приводить к увеличению летальности, однако надо помнить, что чрезмерные дозы витамина D могут повышать уровень FGF-23 [75]. Механизм действия FGF-23 в норме и патологии представлен на рис. 4.
До настоящего времени не разработаны подходы к коррекции уровня FGF-23 у больных с ХПН, однако появились обнадеживающие результаты при применении цинакалцета, который снижал уровень FGF-23 [76, 77], супрессируя функции остеобластов (рис. 5). С другой стороны, применение ингибиторов ангиотензина II приводит к повышению Klotho mRNA [78] и увеличению продолжительности жизни [79].
Литература
- Riminucci M., Collins M. T., Fedarko N. S. et al. FGF-23 in fibrous dysplasia of bone and its relationship to renal phosphate wasting // Journal of Clinical Investigation. 2003; 112 (5): 683–692.
- Khosravi A., Cutler C. M., Kelly M. H. et al. Determination of the elimination half-life of fibroblast growth factor-23 // Journal of Clinical Endocrinology and Metabolism. 2007; 92 (6): 2374–2377.
- Sitara D., Razzaque M. S., Hesse M. et al. Homozygous ablation of fibroblast growth factor-23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice // Matrix Biology. 2004; 23 (7): 421–432.
- Shimada T., Kakitani M., Yamazaki Y. et al. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism // Journal of Clinical Investigation. 2004; 113 (4): 561–568.
- Kuro-o M., Matsumura Y., Aizawa H. et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing // Nature. 1997; 390: 45–51.
- Shimada T., Hasegawa H., Yamazaki Y. et al. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis // J Bone Miner Res. 2004; 19: 429–435.
- Shimada T., Yamazaki Y., Takahashi M. et al. Vitamin D receptor-independent FGF23 actions in regulating phosphate and vitamin D metabolism // Am J Physiol Renal Physiol. 2005; 289: F1088-F1095.
- Saito H., Kusano K., Kinosaki M. et al Human fibroblast growth factor-23 mutants suppress Na+-dependent phosphate co-transport activity and 1 alpha,25-dihydroxyvitamin D3 production // J Biol Chem. 2003, 278: 2206–2211.
- Ben-Dov I. Z., Galitzer H., Lavi-Moshayoff V. et al. The parathyroid is a target organ for FGF23 in rats // J Clin Invest. 2007; 117: 4003–4008.
- Krajisnik T., Bjorklund P., Marsell R. et al. Fibroblast growth factor-23 regulates parathyroid hormone and 1 alpha-hydroxylase expression in cultured bovine parathyroid cells // J Endocrinol. 2007; 195: 125–131.
- Lorenz-Depiereux B., Bastepe M., Benet-Pagиs A. et al. DMP1 mutations in autosomal recessive hypophosphatemia implicate a bone matrix protein in the regulation of phosphate homeostasis // Nat Genet. 2006; 38: 1248–1250.
- Liu S., Tang W., Zhou J. et al. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D // J. Am. Soc. Nephrol. 2006; 17: 1305–1315.
- Nishi H., Nii-Kono T., Nakanishi S. et al. Intravenous calcitriol therapy increases serum concentration of fibroblast growth factor 23 in dialysis patients with secondary hyperparathyroidism // Nephron Clin Pract. 2005; 101: c94-c99.
- Perwad F., Azam N., Zhang M. Y. et al. Dietary and serum phosphorus regulate fibroblast growth factor 23 expression and 1,25-dihydroxyvitamin D metabolism in mice // Endocrinology. 2005; 146: 5358–5364.
- Carrillo-Lуpez N., Romбn-Garcнa P., Rodrнguez-Rebollar A. et al. Indirect regulation of PTH by estrogens may require FGF23 // J Am Soc Nephrol. 2009; 20: 2009–2017.
- Schouten B. J., Hunt P. J., Livesey J. H., Frampton C. M., Soule S. G. FGF23 elevation and hypophosphatemia after intravenous iron polymaltose: a prospective study // J Clin Endocrinol Metab. 2009; 94: 2332–2337.
- Gutierrez O., Isakova T., Rhee E. et al. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease // Journal of the American Society of Nephrology. 2005; 16 (7): 2205–2215.
- Gutierrez O., Isakova T., Rhee E. et al. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease // J Am Soc Nephrol. 2005; 16: 2205–2215.
- Seiler S., Heine G. H., Fliser D. Clinical relevance of FGF-23 in chronic kidney disease // Kidney International. 2009; 114, supplement: S34–S42.
- Gutierrez O., Isakova T., Rhee E. et al. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease // Journal of the American Society of Nephrology. 2005; 16 (7): 2205–2215.
- Koh N., Fujimori T., Nishiguchi S. et al. Severely reduced production of klotho in human chronic renal failure kidney // Biochemical and Biophysical Research Communications. 2001; 280 (4): 1015–1020.
- Imanishi Y., Inaba M., Nakatsuka K. et al. FGF-23 in patients with end-stage renal disease on hemodialysis // Kidney Int. 2004; 65: 1943–1946.
- Nishi H., Nii-Kono T., Nakanishi S. et al. Intravenous calcitriol therapy increases serum concentrations of fibroblast growth factor-23 in dialysis patients with secondary hyperparathyroidism // Nephron Clin Pract. 2005; 101: c94-c99.
- Saito H., Maeda A., Ohtomo S. et al. Circulating FGF-23 is regulated by 1-alpha, 25-dihydroxyvitamin D3 and phosphorus in vivo // J Biol Chem. 2005; 280: 2543–2549.
- Kifor O., Moore F. D. Jr., Wang P. et al. Reduced immunostaining for the extracellular Ca2+-sensing receptor in primary and uremic secondary hyperparathyroidism // J Clin Endocrinol Metab. 1996; 81: 1598–1606.
- Yano S., Sugimoto T., Tsukamoto T. et al. Association of decreased calcium-sensing receptor expression with proliferation of parathyroid cells in secondary hyperparathyroidism // Kidney Int. 2000; 58: 1980–1986.
- Tokumoto M., Tsuruya K., Fukuda K., Kanai H., Kuroki S., Hirakata H. Reduced p21, p27 and vitamin D receptor in the nodular hyperplasia in patients with advanced secondary hyperparathyroidism // Kidney Int. 2002; 62: 1196–1207.
- Komaba H., Goto S., Fujii H. et al. Depressed expression of Klotho and FGF receptor 1 in hyperplastic parathyroid glands from uremic patients // Kidney Int. 2010; 77: 232–238.
- Kumata C., Mizobuchi M., Ogata H. et al. Involvement of α-klotho and fibroblast growth factor receptor in the development of secondary hyperparathyroidism // Am J Nephrol. 2010; 31: 230–238.
- Galitzer H., Ben-Dov I. Z., Silver J., Naveh-Many T. Parathyroid cell resistance to fibroblast growth factor 23 in secondary hyperparathyroidism of chronic kidney disease // Kidney Int. 2010; 77: 211–218.
- Canalejo R., Canalejo A., Martinez-Moreno J. M. et al. FGF23 fails to inhibit uremic parathyroid glands // J Am Soc ephrol. 2010; 21: 1125–1135.
- Nakanishi S., Kazama J. J., Nii-Kono T. et al. Serum fibroblast growth factor-23 levels predict the future refractory hyperparathyroidism in dialysis patients // Kidney Int. 2005; 67: 1171–1178.
- Kazama J. J., Sato F., Omori K. et al. Pretreatment serum FGF-23 levels predict the efficacy of calcitriol therapy in dialysis patients // Kidney Int. 2005; 67: 1120–1125.
- Guillaume Jean, Jean-Claude Terrat, Thierry Vanel et al. High levels of serum fibroblast growth factor (FGF)-23 are associated with increased mortality in long haemodialysis patients // Nephrol. Dial. Transplant. 2009, 24 (9): 2792–2796.
- Mirza M. A., Larsson A., Melhus H., Lind L., Larsson T. E. Serum intact FGF23 associate with left ventricular mass, hypertrophy and geometry in an elderly population // Atherosclerosis. 2009; 207 (2): 546–551.
- Kardami E. et al. Fibroblast growth factor 2 isoforms and cardiac hypertrophy // Cardiovasc Res. 2004; 63 (3): 458–466.
- Negishi K., Kobayashi M., Ochiai I. et al. Association between fibroblast growth factor 23 and left ventricular hypertrophy in maintenance hemodialysis patients. Comparison with B-type natriuretic peptide and cardiac troponin T // Circ J. 2010, Nov 25; 74 (12): 2734–2740.
- Christian Faul Ansel P. Amaral, Behzad Oskouei et al. FGF23 induces left ventricular hypertrophy // J Clin Invest. 2011; 121 (11): 4393–4408.
- Morkin E. Control of cardiac myosin heavy chain gene expression // Microsc Res Tech. 2000; 50 (6): 522–531.
- Izumo S. et al. Myosin heavy chain messenger RNA and protein isoform transitions during cardiac hypertrophy. Interaction between hemodynamic and thyroid hormone-induced signals // J Clin Invest. 1987; 79 (3): 970–977.
- Molkentin J. D. et al. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy // Cell. 1998; 93 (2): 215–228.
- Komuro I., Yazaki Y. Control of cardiac gene expression by mechanical stress // Ann Rev Physiol. 1993; 55: 55–75.
- Rimbaud S. et al. Stimulus specific changes of energy metabolism in hypertrophied heart // J Mol Cell Cardiol. 2009; 46 (6): 952–959.
- Urakawa I. et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23 // Nature. 2006; 444 (7120): 770–774.
- Jaye M., Schlessinger J., Dionne C. A. Fibroblast growth factor receptor tyrosine kinases: molecular analysis and signal transduction // Biochim Biophys Acta. 1992; 1135 (2): 185–199.
- Zhang X., Ibrahimi O. A., Olsen S. K., Umemori H., Mohammadi M., Ornitz D. M. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family // J Biol Chem. 2006; 281 (23): 15694–15700.
- Yu X. et al. Analysis of the biochemical mechanisms for the endocrine actions of fibroblast growth factor-23 // Endocrinology. 2005; 146 (11): 4647–4656.
- Jacques Blacher, Alain P. Guerin, Bruno Pannier et al. Arterial Calcifications, Arterial Stiffness, and Cardiovascular Risk in End-Stage Renal Disease Hypertension. 2001; 38: 938–942.
- Kalpakian M. A., Mehrotra R. Vascular calcification and disordered mineral metabolism in dialysis patients // Semin Dial. 2007; 20: 139–143.
- London G. M. Cardiovascular calcifications in uremic patients: clinical impact on cardiovascular function // Journal of the American Society of Nephrology. 2003; 14 (supplement 4): S305–S309.
- Jono S., McKee M. D., Murry C. E. et al. Phosphate regulation of vascular smooth muscle cell calcification // Circulation Research. 2000; 87 (7): E10–E17.
- Li X., Yang H. Y., Giachelli C. M. Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification // Circulation Research. 2006; 98 (7): 905–912.
- Yang H., Curinga G., Giachelli C. M. Elevated extracellular calcium levels induce smooth muscle cell matrix mineralization in vitro // Kidney International. 2004; 66 (6): 2293–2299.
- Giachelli C. M. Vascular calcification mechanisms // Journal of the American Society of Nephrology. 2004; 15 (12): 2959–2964.
- Nasrallah M. M., El-Shehaby A. R., Salem M. M. et al. Fibroblast growth factor-23 (FGF-23) is independently correlated to aortic calcification in haemodialysis patients // Nephrol Dial Transplant. 2010; 25 (8): 2679–2685.
- Inaba M., Okuno S., Imanishi Y. et al. Role of fibroblast growth factor-23 in peripheral vascular calcification in non-diabetic and diabetic hemodialysis patients // Osteoporos Int. 2006; 17: 1506–1513.
- Giorgio Coen, Paolo De Paolis, Paola Ballanti et al. Peripheral artery calcifications evaluated by histology correlate to those detected by CT: relationships with fetuin-A and FGF-23 // J. Nephrol. 2011; 24 (03): 313–321.
- Coen G., Ballanti P., Silvestrini G. et al. Immunohistochemical localization and mRNA expression of matrix Gla protein and fetuin-A in bone biopsies of hemodialysis patients // Virchows Arch. 2009; 454: 263–271.
- Ketteler M., Wanner C., Metzger T. et al. Deficiencies of calcium-regulatory proteins in dialysis patients: a novel concept of cardiovascular calcification in uremia // Kidney Int Suppl. 2003; 84: 84–87.
- Majd A. I. Mirza, Tomas Hansen, Lars Johansson et al. Relationship between circulating FGF23 and total body atherosclerosis in the community // Nephrol. Dial. Transplant. 2009; 24 (10): 3125–3131.
- Mirza M. A., Larsson A., Lind L. et al. Circulating fibroblast growth factor-23 is associated with vascular dysfunction in the community // Atherosclerosis. 2009; 205 (2): 385–390.
- Eknoyan G., Levin A., Levin N. W. Bone metabolism and disease in chronic kidney disease // Am J Kidney Dis. 2003: 42: 1–201.
- Li Y. C., Kong J., Wei M. et al. 1,25-Dihydroxyvitamin D (3) is a negative endocrine regulator of the renin-angiotensin system // J Clin Invest. 2002: 110: 229–238.
- Li Y. C. Vitamin D regulation of the renin-angiotensin system // J Cell Biochem. 2003: 88: 327–331.
- Tokuda N., Kano M., Meiri H. et al. Calcitriol therapy modulates the cellular immune responses in hemodialysis patients // Am J Nephrol. 2000: 20: 129–137.
- Tabata T., Shoji T., Kikunami K. et al. In vivo effect of 1 alpha-hydroxyvitamin D3 on interleukin-2 production in hemodialysis patients // Nephron. 1988: 50: 295–298.
- Welsh J. Induction of apoptosis in breast cancer cells in response to vitamin D and antiestrogens // Biochem Cell Biol. 1994: 72: 537–554.
- Yamamoto T., Kozawa O., Tanabe K., Akamatsu S., Matsuno H., Dohi S., Hirose H., Uematsu T. 1,25-Dihydroxyvitamin D3 stimulates vascular endothelial growth factor release in aortic smooth muscle cells: Role of p38 mitogen-activated protein kinase // Arch Biochem Biophys. 2002: 398: 1–6.
- Xiang W., Kong J., Chen S. et al. Cardiac hypertrophy in vitamin D receptor knockout mice: Role of the systemic and cardiac renin-angiotensin systems // Am J Physiol Endocrinol Metab. 2005: 288: E125–E132.
- Ravani P., Malberti F., Tripepi G. et al. Vitamin D levels and patient outcome in chronic kidney disease // Kidney International. 2009; 75 (1): 88–95.
- Zittermann A., Schleithoff S. S., Koerfer R. Vitamin D insufficiency in congestive heart failure: Why and what to do about it? // Heart Fail Rev. 2006; 11: 25–33.
- Zittermann A., Schleithoff S. S., Gotting C. et al. Poor outcome in end-stage heart failure patients with low circulating calcitriol levels // Eur J Heart Fail. 2008: 10: 321–327.
- Pilz S., Marz W., Wellnitz B. et al. Association of vitamin D deficiency with heart failure and sudden cardiac death in a large cross-sectional study of patients referred for coronary angiography // J Clin Endocrinol Metab. 2008; 93: 3927–3935.
- Nishi H., Nii-Kono T., Nakanishi S. et al. Intravenous calcitriol therapy increases serum concentrations of fibroblast growth factor-23 in dialysis patients with secondary hyperparathyroidism // Nephron Clin Pract. 2005; 101 (2): c94–99.
- James B. Wetmore, Shiguang Liu, Ron Krebill et al. Effects of Cinacalcet and Concurrent Low-Dose Vitamin D on FGF23 Levels in ESRD. CJASN January 2010, vol. 5, № 1: 110–116.
- Hryszko T., Brzosko S., Rydzewska-Rosolowska A. et al. Cinacalcet lowers FGF-23 level together with bone metabolism in hemodialyzed patients with secondary hyperparathyroidism // Int Urol Nephrol Int Urol Nephrol. 2011: 27.
- Tang R., Zhou Q., Shu J. et al. Effect of cordyceps sinensis extract on Klotho expression and apoptosis in renal tubular epithelial cells induced by angiotensin II // Zhong Nan Da Xue Xue Bao Yi Xue Ban. 2009; 34: 300–307.
- Kurosu H., Yamamoto M., Clark J. D. et al. Suppression of aging in mice by the hormone Klotho // Science. 2005; 309: 1829–1833.
Статья опубликована в журнале Лечащий Врач
материал MedLinks.ru






