Болезнь Бурневилля–Прингля
Л. А. Юсупова, доктор медицинских наук
З. Ш. Гараева, кандидат медицинских наук, доцент
Е. И. Юнусова, кандидат медицинских наук, доцент
Г. И. Мавлютова, кандидат медицинских наук
Л. А. Хаертдинова, кандидат медицинских наук, доцент
Э. Э. Галиханова, кандидат медицинских наук
В. Н. Рокицкая, кандидат медицинских наук, доцент
ГБОУ ДПО КГМА Минздравсоцразвития России, Казань
Болезнь Бурневилля–Прингля (туберозный склероз) — гетерогенное, генетически детерминированное заболевание, характеризующееся гиперплазией производных экто- и мезодермы, поражением кожи, нервной системы и наличием доброкачественных опухолей (гамартом) в различных органах. В 1880 г. описание болезни опубликовано Д. М. Бурневиллем и в 1890 г. Дж. Дж. Принглом.
Болезнь Бурневилля–Прингля наследуется по аутосомно-доминантному типу, отличается варьирующей экспрессивностью и неполной пенетрантностью. Развитие болезни определяется сцеплением с локусами 9q34 (первого типа — TSC1), Ilql4—llq23 и 16р13.3 (второго типа — TSC2). Имеются данные о наличии мутации гена на 12-й хромосоме. Предполагается, что гамартин (кодируется TSC1) является белком, подавляющим рост опухолей, а туберин (кодируется TSC2) регулирует эндоцитоз. Возможен дефект в системе репарации ДНК, о чем свидетельствует повышенная чувствительность клеток к ионизирующей радиации. В 50–75% случаев заболевание может быть обусловлено новыми мутациями. Частота болезни Бурневилля–Прингля составляет 1:30 000 населения. Распространенность среди новорожденных варьирует до 5–7 случаев на 100 000 новорожденных [1, 2].
В ангиофибромах наблюдается разрастание соединительной ткани, пролиферация мелких сосудов, преимущественно капиллярного типа, расширение их просветов. Соединительнотканные невусы при болезни Бурневилля–Прингля представлены коллагеномами. Эпителий обычно без особенностей, но может быть изменен по типу эпидермального невуса. Дерма утолщена за счет гипертрофированных коллагеновых волокон [1].
Клинические симптомы болезни Бурневилля–Прингля появляются в первые годы жизни, но могут существовать с рождения. Процесс постепенно прогрессирует, особенно в период полового созревания. Кожа поражена в 96% случаев [3]. Кожные проявления болезни Бурневилля–Прингля представлены ангиофибромами и фиброматозными очагами на лице, околоногтевыми фибромами, шагреневыми бляшками, гипомеланотическими пятнами, пигментными пятнами цвета «кофе с молоком». В 1998 г. были приняты диагностические критерии заболевания (табл.) [4].
По приведенным диагностическим критериям несомненный диагноз болезни Бурневилля–Прингля ставится в случае двух или одного первичного признака и двух вторичных признаков. Возможный диагноз: один первичный признак и один вторичный признак. Предположительный диагноз: или один первичный признак, или два (и более) вторичных признака (табл.).

Гипопигментированные пятна на коже существуют с рождения или появляются в грудном возрасте и являются одним из наиболее частых кожных проявлений болезни Бурневилля–Прингля. На первом году жизни их находят у 80% больных, на втором — у 100% [3, 5]. С возрастом наблюдается тенденция к увеличению их числа. Гипопигментные пятна при этом заболевании локализуются преимущественно на туловище и ягодицах. Характерной их особенностью является асимметричность расположения. Отмечена вариабельность числа, размера и формы пятен. Наиболее характерные из них имеют очертания листа, заостренного с одной стороны и закругленного с другой, бледно-сероватой или молочно-белой окраски. На светлой коже их видно только с помощью лампы Вуда. С течением времени пятна могут медленно репигментироваться. Диагностическое значение имеют только множественные элементы, особенно при сочетании их с эпилептиформными припадками. С младенчества могут выявляться белые пряди волос, ресниц и бровей, которые, как и гипопигментные пятна, являются характерным признаком болезни Бурневилля–Прингля.
Наряду с гипопигментными пятнами при болезни Бурневилля–Прингля в 15,4% случаев встречаются пигментные пятна цвета «кофе с молоком», которые не отличается от таковых у здоровых лиц, но наличие их в сочетании с другими симптомами помогает в постановке диагноза.
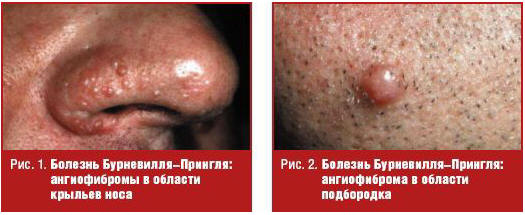
В 47–90% случаев наблюдаются ангиофибромы, являющиеся облигатным признаком болезни Бурневилля–Прингля. Ангиофибромы на первом году жизни появляются только у 20% больных, к трем годам — у 50%, располагаются, как правило, симметрично на крыльях носа, в носогубных складках и на подбородке. Они представляют собой мелкие полушаровидные плотноватые опухолевидные элементы, величиной 1–5 мм, телесного, желтовато-красного или красновато-коричневатого цвета. Их поверхность блестящая, гладкая, но может быть веррукозной, покрытой телеангиэктазиями (рис. 1, 2).
В области кожи лба, волосистой части головы, щек наблюдающиеся крупные опухолевидные фиброматозные очаги также являются облигатным признаком заболевания и встречаются у 25% больных болезнью Бурневилля–Прингля. Фиброматозные очаги могут быть как одиночными, так и множественными, имеют вариабельную окраску — от цвета нормальной кожи до светло-коричневого, несколько выступают над окружающей кожей, мягкой или плотноватой консистенции. Они часто появляются уже на первом году жизни и являются одним из первых клинических симптомов заболевания. Фиброзные бляшки чаще всего локализуются на лбу. Размеры и число их могут варьировать. Мягкие фибромы встречаются у 30% больных, представляют собой множественные или единичные мягкие образования на ножках, мешотчатой формы, растущие на шее, туловище и конечностях. Другой вариант мягких фибром представляет собой множественные, несколько приподнятые над поверхностью кожи (и такого же цвета) мелкие образования, размером меньше 0,3 см в диаметре, располагающиеся на туловище и шее и напоминающие гусиную кожу.
Часто встречаются подногтевые и околоногтевые фибромы (опухоли Кенена), гипертрофические изменения десен. Околоногтевые фибромы, являющиеся облигатным признаком болезни Бурневилля–Прингля, представляют собой тусклые, красные либо мясного цвета папулы или узлы, растущие от ногтевого ложа или вокруг ногтевой пластинки. Опухоль Кенена появляется в позднем детском возрасте и встречается в 17–52% случаев. В большинстве случаев околоногтевые фибромы появляются на втором десятилетии жизни. Наиболее часто они локализуются на ногах. Размер их варьирует от 1 мм до 1 см в диаметре. Гистологически опухоль Кенена представляет собой ангиофиброму.
Шагреневидные бляшки, или «шагреневая кожа», развиваются в первое десятилетие жизни примерно у 40% больных и представляют собой соединительнотканные невусы. В большинстве случаев «шагреневая кожа» появляется на втором десятилетии жизни. Шагреневидные бляшки могут быть как единичными, так и множественными, от мелкого размера до 10 и более см в диаметре, с пористой поверхностью типа «лимонной корки». Участки «шагреневой кожи» наблюдаются преимущественно в пояснично-крестцовой области, имеют вид плоских, слегка возвышающихся очагов, располагающихся преимущественно в люмбосакральной области, цвета нормальной кожи или слабо пигментированные.
При болезни Бурневилля–Прингля встречаются разнообразные системные изменения организма. Неврологические симптомы могут быть самыми первыми признаками болезни, внезапно проявляющимися на фоне внешнего здоровья и благополучия у ребенка без заметных дисплазий и нарушений развития. Возникают они в возрасте 3–4 месяцев в виде судорожных приступов. Поражения нервной системы являются доминирующими в клинической картине болезни Бурневилля–Прингля. Наиболее характерны судорожные пароксизмы, умственная отсталость, нарушения поведения, изменения в цикле «сон–бодрствование».
Судорожные пароксизмы — один из наиболее значимых симптомов болезни Бурневилля–Прингля — наблюдаются у 80–92% больных [6] и чаще всего являются манифестным симптомом заболевания. Первые приступы бывают общими тоническими, затем они становятся полиморфными (общие, фокальные, большие, малые, кивки, закатывание глаз, замирания, судороги). Частые припадки в основном наблюдаются до 6–7-летнего возраста, а затем они могут пройти. У ряда больных приступы продолжаются и в более старшем возрасте. У некоторых они имеют тяжелое течение, может развиться эпилептический статус с летальным исходом [7]. Чем раньше начинается эпилепсия, тем тяжелее умственная отсталость [3]. Эпилептические пароксизмы при болезни Бурневилля–Прингля нередко резистентны к противосудорожной терапии, могут приводить к развитию нарушений интеллекта и поведения и являются одной из главных причин инвалидности у детей с болезнью Бурневилля–Прингля. Среди факторов, детерминирующих резистентность к противосудорожной терапии, наибольшее значение имеют: дебют в возрасте до одного года, наличие нескольких типов приступов, высокая частота приступов, изменение характера приступов с течением заболевания [8].
Наиболее типичными поражениями головного мозга при болезни Бурневилля–Прингля являются корковые туберы, субэпендимальные узлы и аномалии белого вещества мозга. Кальцификация туберов отмечается в 54% случаев и увеличивается с возрастом больных. Большую значимость в верификации туберов при обследовании больных болезнью Бурневилля–Прингля имеет магнитно-резонансная томография (МРТ), которая позволяет визуализировать туберы в 95% случаев.
Субэпендимальные узлы встречаются в 95% случаев и выявляются как при компьютерном томографическом (КТ), так и при МРТ-исследованиях мозга. Субэпендимальные узлы в большинстве случаев множественные, прилегающие друг к другу. Локализуются, как правило, в стенках боковых желудочков, реже — в стенках III и IV желудочков мозга. Субэпендимальные узлы нередко трансформируются в гигантоклеточную астроцитому и выявляются у 10–15% больных [5]. Субэпендимальные гигантоклеточные астроцитомы манифестируют обычно между 5-м и 10-м годами жизни (средний возраст в момент выявления опухоли — 13 лет), как правило, имеют тенденцию к росту и всегда локализуются у межжелудочкового отверстия. У 10% больных при болезни Бурневилля–Прингля описаны поражения мозжечка.
Среди более редких неврологических симптомов встречаются центральные спастические параличи, пирамидные и экстрапирамидные симптомы, при росте опухоли в полость желудочков — внутренняя гидроцефалия. Могут быть обнаружены застойные соски зрительных нервов, их атрофия, в редких случаях — эндокринные расстройства в виде адреногенитального синдрома, нарушения со стороны черепно-мозговых нервов. В редких случаях наблюдаются спонгиобластомы, развивающиеся из очага болезни Бурневилля–Прингля, с соответствующей симптоматикой опухоли.
Развитие умственной отсталости замечается позже появления судорожного синдрома и регистрируется примерно у 49% больных, постепенно усугубляется вследствие деструкции мозга, пораженного болезнью Бурневилля–Прингля, и может достигать степени глубокой имбецильности. Отстает психическое развитие, разрушаются моторные навыки, нарушается речь. К пубертатному периоду нарушение интеллекта может достигать степени идиотии. Д. М. Бурневиллем (1880) описание этой болезни было опубликовано под названием «вклад в изучение идиотии». Однако при нерезко выраженном деструктивном процессе клинические проявления не столь тяжелы, эволютивная динамика развития нервной системы в известной степени перекрывает патологический процесс, и к психиатру этих детей приводят с олигофреническим интеллектуальным недоразвитием [3, 7].
Интеллектуальный дефект может резко углубляться при развитии психотических нарушений. Наблюдаются шизофреноподобные психозы со страхами, манией преследования, аномалии поведения с психопатическими чертами, изменениями личности по ограниченному типу с вязкостью, некритичностью, назойливостью. Даже при легких формах слабоумия, когда дефект нарастает медленно, годами, приходится помнить о том, что это заболевание имеет прогредиентное течение и, следовательно, неблагопрятный прогноз. Однако у 30% больных не отмечают слабоумия. Но статистика не точна, так как приводятся данные по регистрации обратившихся больных.
Нередко при поражении глаз выявляют застойные соски, иногда атрофию зрительных нервов. При офтальмоскопическом обследовании более чем у 50% больных наблюдается патогномоничная картина ретинальной факомы. Эти невоидные образования бывают трех типов. При первом, наиболее распространенном варианте гамартомы имеют нежную, относительно плоскую и гладкую поверхность, оранжево-розовый цвет, округлую или овальную форму, локализуются преимущественно в поверхностных слоях сетчатки. При втором — гамартомы имеют узловатый вид и напоминают тутовую ягоду. Они белого цвета, кальцифицированные, светонепроницаемые. При третьем варианте гамартомы сочетают в себе признаки первых двух. Они имеют округлую форму с узловатым и кальцифицированным центром и полупрозрачной, гладкой периферией оранжево-розового цвета. Такие проявления имеют важное диагностическое значение из-за характерного вида. У некоторых больных это может быть одним из единственных проявлений болезни Бурневилля–Прингля. Значительно реже регистрируются другие изменения органа зрения: хориоретинит, зоны депигментации, врожденная катаракта, врожденная слепота. Встречаются также соединительнотканные узелки на конъюнктиве. Клинические проявления гамартом наблюдаются крайне редко. Основным симптомом является прогрессирующее снижение зрения [2, 7].
Опухоли во внутренних органах у многих больных не вызывают клинических симптомов, но часто обнаруживаются на аутопсии, особенно опухоли почек. Полагают, что опухоли почек выявляются у 40–50% больных. Это множественные билатериальные мелкие гамартомы из соединительнотканных волокон, жировой ткани, эпителия. Иногда встречаются крупные опухоли почек.
Изменения сердечно-сосудистой системы при болезни Бурневилля–Прингля проявляются развитием рабдомиом, которые нередко служат первым клиническим признаком болезни Бурневилля–Прингля наряду с гипопигментными пятнами. В 1863 г. Реклингхаузен описал сочетание поражения мозга с рабдомиомой. Рабдомиомы встречаются в 30–60% случаев и выявляются чаще у лиц мужского пола (соотношение 2:1). Наиболее высокая частота рабдомиомы сердца при болезни Бурневилля–Прингля наблюдается у новорожденных и детей грудного возраста. Рабдомиомы сердца, как правило, быстро увеличиваются во время второй половины беременности, в основном достигают максимальных величин к моменту рождения, а затем постепенно уменьшаются в размерах. Большинство рабдомиом исчезают бесследно. Спонтанная регрессия рабдомиом может быть у детей младше шести лет. После шести лет опухоли обычно не исчезают, однако могут несколько уменьшаться в размере. Регресс опухолей может наблюдаться как в размере, так и в их числе [5].
Не являются большой редкостью и поражения легких в виде фиброзных опухолей, фибролейомиом, кистозных образований, интерстициального фиброза. У больных возникают приступы диспноэ, возвратный спонтанный пневмоторакс, кровохаркание, легочная недостаточость. Описываются опухоли поджелудочной железы, печени, мочевого пузыря, желудочно-кишечного тракта и других органов. На слизистых оболочках встречаются фибромы десны, языка, глотки, гортани [7].
Дифференциальную диагностику при болезни Бурневилля–Прингля гипопигментированных пятен следует проводить с очаговой формой витилиго, анемическим невусом, отрубевидным лишаем, беспигментным невусом, послевоспалительной гипопигментацией. Ангиофиброму следует дифференцировать с трихолеммомой, сирингомой, внутридермальным невоклеточным невусом. Опухоль Кенена следует дифференцировать с простыми бородавками [3].
При диагностике кожных поражений не требуется дополнительных параклинических исследований, если больной обращается с псевдоаденомами и другими типичными хорошо видимыми поражениями. Они настолько патогномоничны, что в рамках рутинной диагностики обычно нет необходимости прибегать к патоморфологическому исследованию. Но в то же время, зная о широком спектре проявления болезни Бурневилля–Прингля, нельзя останавливаться только на уровне дерматологической диагностики. Больного необходимо направить к психиатру, невропатологу, окулисту, терапевту, хирургу. Необходимо сделать электрокардиограмму, рентгенографию грудной клетки, черепа, кистей и стоп, электроэнцефалографию, КТ, МРТ, анализ мочи (гематурия при поражении почек). Может возникнуть потребность в осмотре кожи лампой Вуда при предположении о слабых проявлениях у родственников больных неясных белых пятнах. Можно сделать и патоморфологическое исследование белых пятен, если кожная симптоматика представлена только ахромическими поражениями. Лампу Вуда целесообразно использовать также при обследовании детей, родившихся oт родителей, больных болезнью Бурневилля–Прингля. Это исследование особенно важно в том возрасте, когда еще типичные псевдоаденомы отсутствуют (до 3–5 лет). При тяжелой форме заболевания 30% больных не доживают до 5 лет; 50–75% умирают в детском и подростковом возрасте. Нередки злокачественные глиомы. Обязательно следует проводить медико-генетическое консультирование [3].
Прогноз для выздоровления неблагоприятный. При тяжелых системных изменениях высока летальность в детском и молодом возрасте от эпилептического статуса, сердечной, почечной или легочной недостаточности. Выраженность кожных изменений не влияет на риск вовлечения в процесс внутренних органов.
При лечении наиболее крупные элементы удаляют электрокоагуляцией, криодеструкцией, лазерным излучением. Наблюдается уменьшение размеров ангиофибром от Тигазона (по 1 мг на кг массы тела) [1]. Может быть полезна дермабразия, которую следует проводить после стабилизации процесса. Длительно назначают антиконвульсивные препараты (Дифенин и др.), периодически — средства, снижающие внутричерепное давление, нормализующие сердечный ритм при рабдомиоме сердца. Терапия выбора при опухолях головного мозга — хирургическое удаление. С целью пренатальной диагностики может быть использована эхокардиография для выявления у плода рабдомиомы сердца.
Литература
- Мордовцев В. Н., Мордовцева В. В., Мордовцева В. В. Наследственные болезни и пороки развития кожи. Атлас. М.: Наука, 2004. С. 40–42.
- Страхова О. С., Катышева О. В., Дорофеева М. Ю., Перминов В. С., Пивоварова А. М., Осипова Э. К., Добрынина М. В., Чумак О. И. Туберозный склероз // Российский медицинский журнал. 2004. № 3. С. 52–54.
- Фицпатрик Т., Джонсон Р., Вулф К., Полано М., Сюрмонд Д. Дерматология: атлас-справочник. 1999. С. 460–466.
- Roach E. S., DiMario F. J., Kandt R. S., Northrup H. Tuberous Sclerosis Consensus Conference: Recommendations for Diagnostic Evaluation // Journal of Child Neurology. 1999. V. 14. Р. 401–407.
- Дорофеева М. Ю. Туберозный склероз у детей // Российский вестник перинатологии и педиатрии. 2001. № 4. С. 33–41.
- Curatolo P., Seri S. Seizures. In: Nuberous Sclerosis complex: from Basic Science to Clinical Phenotypes. Ed: Curatolo P. London, England: Mac Keith Press. 2003. Р. 46–77.
- Суворова К. Н., Куклин В. Т., Рукавишникова В. М. Детская дерматовенерология. Казань, 1996. С. 50–56.
- Curatolo P. Tuberous Sclerosis. In: Infantile Spasms and West Syndrome. Ed. by O. Dulac, H. T. Chugani, B. Dalla Bernandina. W. B. Saunders. Company Ltd, London, Philadelphia, Toronto, Sydney, Tokio. 1994. Р. 192–202.
Статья опубликована в журнале Лечащий Врач
материал MedLinks.ru